Yinan Zheng, Brian T. Joyce, Elena Colicino, Lei Liu, Wei Zhang, Qi Dai, Martha J. Shrubsole, Warren A. Kibbe, Tao Gao, Zhou Zhang, Nadereh Jafari, Pantel Vokonas, Joel Schwartz, Andrea A. Baccarelli, Lifang Hou
DOI: http://dx.doi.org/10.1016/j.ebiom.2016.02.008
Highlights
-
We prospectively examined blood Δage and its ability to predict cancer risks.
-
Epigenetic age older than chronological age elevated cancer risk.
-
Δage predicted cancer incidence and mortality in a dose-responsive manner.
-
The Δage–cancer relationship was a nonlinear ‘J-shape’ for subjects measured before 2003. Blood-based epigenetic age is a potential biomarker for cancer early detection.
This study studies a way to calculate your body's age, not based on how old you are, but by measuring a number of markers in your blood — called epigenetic age. This paper also looks at how good epigenetic age over time is at predicting whether you'll get cancer, and whether you'll die from it. The authors found that epigenetic age could be good at predicting both of these things, which means that someday it could be developed into a blood test for diagnosing cancer, and for helping patients figure out how long they'll live.
Abstract
Biological measures of aging are important for understanding the health of an aging population, with epigenetics particularly promising. Previous studies found that tumor tissue is epigenetically older than its donors are chronologically. We examined whether blood Δage (the discrepancy between epigenetic and chronological ages) can predict cancer incidence or mortality, thus assessing its potential as a cancer biomarker. In a prospective cohort, Δage and its rate of change over time were calculated in 834 blood leukocyte samples collected from 442 participants free of cancer at blood draw. About 3–5 years before cancer onset or death, Δage was associated with cancer risks in a dose-responsive manner (P = 0.02) and a one-year increase in Δage was associated with cancer incidence (HR: 1.06, 95% CI: 1.02–1.10) and mortality (HR: 1.17, 95% CI: 1.07–1.28). Participants with smaller Δage and decelerated epigenetic aging over time had the lowest risks of cancer incidence (P = 0.003) and mortality (P = 0.02). Δage was associated with cancer incidence in a ‘J-shaped’ manner for subjects examined pre-2003, and with cancer mortality in a time-varying manner. We conclude that blood epigenetic age may mirror epigenetic abnormalities related to cancer development, potentially serving as a minimally invasive biomarker for cancer early detection.
1. Introduction
As the population continues to age in the coming decades, the need for biological measures of age and more precise screening tests for age-related diseases will become increasingly urgent. Genetic and epigenetic studies have added to the potential clinical utility of this subject area, with researchers identifying elements associated with age-related processes (e.g., telomeres) that nonetheless have unverified predictive power in human populations (Brooks-Wilson, 2013). Epigenetics serves as an intersection between genetic and environmental risk factors for aging processes and age-related diseases, holding great promise for constructing biological age measures that will provide clinical diagnostic tools for aging-related diseases such as cancer. Blood-based epigenetic markers are particularly well-suited to these purposes due to their minimally invasive method of collection and cost-effectiveness on the population scale.
Epigenetic age is a recently developed algorithm that uses DNA methylation measurements to describe biological age at the level of human tissues, cells, and organs (Hannum et al., 2013, Horvath, 2013, Weidner et al., 2014). Epigenetic age does not always parallel chronological age, particularly in tumor samples (Hannum et al., 2013, Horvath, 2013), a discrepancy we refer to here as Δage. Furthermore, since the methods for measuring epigenetic age incorporate loci in pathways related to both cancer development and aging in general (e.g., DNA damage, cellular proliferation, and oxidative stress) (Hannum et al., 2013, Horvath, 2013), it is highly possible that Δage can be a predictive biomarker for cancer risk, metastasis, and mortality in addition to serving as an indicator of aging. With further study and refinement, the concept of epigenetic age may also be useful for improving our understanding of mechanisms by which age and cancer are related. However, no longitudinal analysis has yet evaluated how blood epigenetic age changes over time prior to cancer diagnosis or cancer-related death, and whether blood Δage can predict future risk of cancer incidence and mortality.
Our objective is to assess whether white blood cell (WBC) Δage can predict cancer incidence and mortality, and to evaluate these predictions over time. We will achieve our goals by comparing multiple estimates of Δageobtained using blood DNA samples collected prior to cancer incidence and death in: 1) individuals who developed cancer relative to cancer-free individuals and 2) individuals who died of cancer relative to both cancer survivors and cancer-free individuals.
2. Materials and Methods
2.1. Study Design and Participants
The U.S. Department of Veterans Affairs' Normative Aging Study (NAS) is a longitudinal cohort established in 1963 (Bell et al., 1966). Between March 1999 and December 2013, 802 out of 829 (96.7%) of active participants agreed to donate blood, 686 of whom were randomly selected and profiled using the Illumina 450 K BeadChip array at up to three follow-up visits separated by median time intervals of 3.5 years (IQR 3.1–5.7) (Supplementary material, Fig. S1). We excluded 18 participants who were non-White or had missing information on race to minimize potential confounding effects of genetic ancestry, 182 participants who had been diagnosed with malignant cancer prior to the first blood draw, and 44 participants who had been diagnosed with neoplasms of uncertain behavior, leaving a total of 442 participants for analysis. Cancer diagnoses and comorbidities (prevalent diabetes, hypertension, coronary artery disease, and stroke) were obtained from questionnaires and confirmed via blinded medical record review. Official death certificates were reviewed by a physician, and all contributing causes coded by an experienced research nurse. Telomere length was measured using quantitative real-time polymerase chain reaction and reported in relative units, as previously described (Hou et al., 2015). Of the 442 participants free of cancer at first blood draw, 132 developed cancer during follow-up (38 prostate, 50 skin, 44 other) and 34 died from cancer (4 prostate, 2 skin, 28 other). The remaining 98 participants died from non-cancer causes or were still alive as of latest follow-up, and are defined as cancer survivors for our analysis (Supplementary material, Fig. S2). Median follow-up time (from first blood draw) to cancer incidence or censoring was 10.1 years (IQR 5.8–12.7) and 11.9 years (IQR 8.8–13.0) to cancer mortality or censoring. This study was approved by the Institutional Review Boards of all participating institutions, and all participants provided written consent.
2.2. Illumina Infinium HumanMethylation450 BeadChip DNA Methylation Profiling
DNA was extracted from the buffy coat using the QIAamp DNA Blood Kit (QIAGEN, Valencia, CA, USA). A total of 500 ng of DNA was used to perform bisulfite conversion using the EZ-96 DNA Methylation Kit (Zymo Research, Orange, CA, USA). To limit chip and plate effects a two stage, age-stratified algorithm was used to randomize samples and ensure similar age distributions across chips and plates. We randomized 12 samples (sampled across all age quartiles) per chip, and then randomized chips to plates. Quality control analysis was performed to remove samples where >1% of probes had a detection P > 0.05. The remaining samples were preprocessed using Illumina-type background correction without normalization as re-implemented in the Bioconductor minfi package and used to generate beta values (Aryee et al., 2014). The working set included all 485,512 CpG and CpH probes for epigenetic age estimation.
2.3. Epigenetic Age and Δage Calculation
We estimated epigenetic age as measured by Hannum's 71-CpG method using Horvath's online calculator (http://labs.genetics.ucla.edu/horvath/dnamage/), which incorporates both previously-published methods (Hannum et al., 2013, Horvath, 2013) based on prior work showing superior predictive accuracy in blood (Marioni et al., 2015). We selected Hannum's method because the model was trained using blood samples and it yielded accurate and longitudinally stable epigenetic age estimation using our blood-based methylation data (Supplementary material, Fig. S3). To remove potential confounding due to alterations in WBC composition with age (Marioni et al., 2015) and naïve T cell changes due to thymic involution (Taub and Longo, 2005), cell abundances were estimated via previously published methods (Horvath, 2013, Houseman et al., 2012). We then used the residuals from a linear regression of epigenetic age on chronological age and these cell type abundances to calculate Δage (with Δage > 0 indicating epigenetic age older than chronological age), resulting in a construct less sensitive to normal age-related changes in WBC composition and immunosenescence (Supplementary material, Fig. S4).
2.4. Statistical Analysis
We performed descriptive analyses of participant characteristics (including epigenetic age and Δage) at first blood draw using linear mixed-effect models. We then expanded these descriptive analyses to assess dose–response via a multivariable linear mixed-effect model comparing Δage across samples (estimated by least squares means) in participants who died from cancer, cancer survivors, and cancer-free participants. Next, we used Random Matrix Theory (Plerou et al., 2002) available in R package isva (Teschendorff et al., 2011) to determine that there were two significant components of variation in our data. We made a heat map (Supplementary material, Fig. S5) of P-values matrix of associations between the significant components and: biological (Houseman's six blood cell type proportions; Houseman et al., 2012), epidemiological (the 11 covariates we considered) and technical factors (plate number, chip number, row number and column number of the chip). We found that the components were strongly associated with biological and technical factors, but not epidemiological factors. Therefore, all subsequent models adjusted for the top two principle components computed from the 450 K array data to account for batch effects and residual biological confounding. We included the third top PC to better capture latent population stratification of the data (Barfield et al., 2014).
Prior to running our survival analyses, we evaluated the proportional hazards assumption for both cancer incidence and mortality by plotting Schoenfeld residuals over time. Although the hazards for cancer incidence were proportional across all follow-up time, the mortality analysis violated the proportional hazards assumption with marginal statistical significance (P = 0.04; data available upon request). Therefore, our subsequent analyses examined Δage and cancer both across all follow-up visits and stratified by year of blood DNA collection. We selected a stratification point of January 1st, 2003 as 1) it separated two data collection ‘cycles’ in the NAS rotation protocol (Supplementary material, Fig. S1), and 2) the resulting subgroups were of similar size. Subsequent testing of the proportional hazards assumption within these strata revealed no violations for either cancer incidence or mortality.
We plotted KM curves, comparing participants epigenetically older than their chronological age (Δage > 0) to those epigenetically younger than their chronological age (Δage ≤ 0) using log-rank tests. Next, we used time-dependent Cox proportional hazards models to evaluate associations between Δage and times to cancer diagnosis and death. We investigated potential nonlinearity of these associations using pointwise HR curves (Meira-Machado et al., 2013) and generalized additive Cox models using penalized splines (Hastie and Tibshirani, 1995). Finally, using the 299 participants with two or more samples, we computed the slope of Δageover time and categorized these participants as having accelerated (Δage slope > 0) or decelerated (Δage slope ≤ 0) epigenetic aging over time. We then used a second set of KM survival curves and log-rank tests to analyze the relationship between Δage, epigenetic aging over time, and cancer.
Our multivariable models additionally adjusted for chronological age at first blood draw, BMI, education, smoking status, pack-years of smoking, and alcohol consumption. One participant with missing data on education was excluded from multivariable analysis. Five other variables (telomere length and presence of four comorbidities) were examined in separate sensitivity analyses of our Cox models. The inclusion of these variables did not substantively affect our estimates (Supplementary material, Table S1) and they were therefore excluded due to having high proportions of missing data. We conducted all analyses using R v3.0.2, two-sided tests, and a statistical significance threshold of P = 0.05.
2.5. Data Availability
The NAS data are available at dbGaP under the accession numbers phs000853.v1. p1 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000853.v1.p1).
2.6. Funding Source
This study was funded by the Epidemiology Research and Information Center, U.S. Department of Veterans Affairs; NIEHS R01-ES015172 . Additional funding support was provided by the Northwestern University Robert H. Lurie Comprehensive Cancer Center Rosenberg Research Fund. The funding institutions had no role in the study design, data collection or analysis, interpretation of findings, manuscript preparation, or decision to pursue peer-review publication. No author has been paid to write this article by a pharmaceutical company or other agency.
3. Results
Characteristics of participants at first blood draw were similar to previous reports (Hou et al., 2015, Zhu et al., 2011), indicating successful randomization. No participant characteristics were associated with Δage(Supplementary material, Table S2). Mean epigenetic age was almost identical to chronological age but with greater variance (Table 1). There were 422 observations of 370 participants during the first half of the follow-up period (pre-2003) and 412 observations of 306 participants during the second half (2003–2013). In the pre-2003 stratum the median times to cancer diagnosis and death were 5.1 (IQR (interquartile range) 2.7–8.6) and 7.0 years (IQR 5.3–10.1), respectively. In the 2003–2013 stratum the median times to cancer diagnosis and death were 3.0 (IQR 0.6–5.7) and 4.6 years (IQR 2.4–7.0), respectively. The pre-2003 analysis included 370 and 52 samples from participants at their first and second time-point of measures, respectively. The 2003–2013 analysis included 72, 247, and 93 samples from participants at their first, second, and third time-point of measures, respectively.

Using data gathered from 2003 to 2013, our dose–response analysis identified greater Δage in cancer survivors and participants who died from cancer (Δage = 0.5 and 2.2 years, respectively) relative to cancer-free participants (Δage = −0.4 years, test for trend P = 0.02) (Fig. 1); Kaplan–Meier (KM) curves comparing Δage also suggested participants with greater Δage had a higher risk of cancer incidence (log-rank P = 0.002) and cancer mortality (log-rank P = 0.04) (Supplementary material, Fig. S6). We observed weaker, non-significant results in analyses using samples before 2003 and using all samples combined.
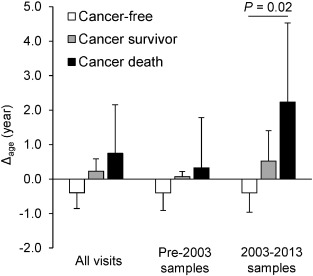
Fig. 1
Dose–response analysis of Δage by cancer status.
Least squares means of Δage across cancer status groups were estimated by linear mixed-effect models to account for repeated measures. We compared participants who died from cancer, cancer survivors, and cancer-free participants and found that severity of cancer outcome increased as Δage increased. Error bars represent standard errors.
Adjusted for covariates, the 2003–2013 time-dependent Cox models revealed that each one-year increase in Δage was associated with greater risks of cancer incidence (HR: 1.06, 95% CI: 1.02–1.10), and mortality (HR: 1.17, 95% CI: 1.07–1.28). We found no associations between Δage and cancer risk in either all samples combined or the pre-2003 samples (Table 2). Point-wise HR curves of Δage illustrated a J-shaped relationship in cancer incidence for samples taken pre-2003 (P = 0.03 for test of nonlinearity) (Fig. 2A ). The risk of cancer mortality increased linearly with Δage in the 2003–2013 samples, but this association was substantially weaker for pre-2003 samples (Fig. 2B). KM survival curves examining Δage rate of change over time showed that participants who were epigenetically young relative to their chronological age (Δage ≤ 0) and had decelerated or stable epigenetic aging over time (Δage slope ≤ 0) had the lowest risk of cancer incidence and death (log-rank P = 0.003 and P = 0.02, respectively, Fig. 3).
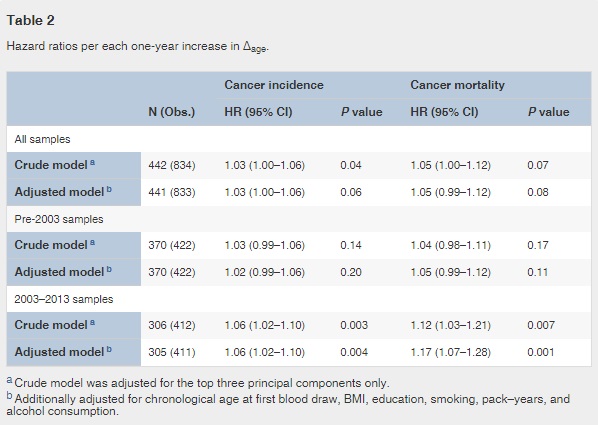
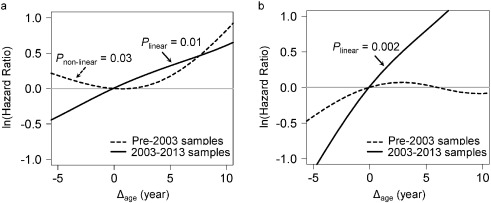
Fig. 2
Point-wise hazard ratio curves between Δage and risk of cancer.
Risk of cancer incidence (A) and mortality (B) represented by log-transformed HR were plotted against Δage in the pre-2003 (black dash line) and 2003–2013 (black solid line) strata. Log-transformed HR = 0 (gray solid line) is equivalent to hazard ratio = 1. Both linear and non-linear relationships were tested using spline analysis. Only significant relationships were noted in the figure.
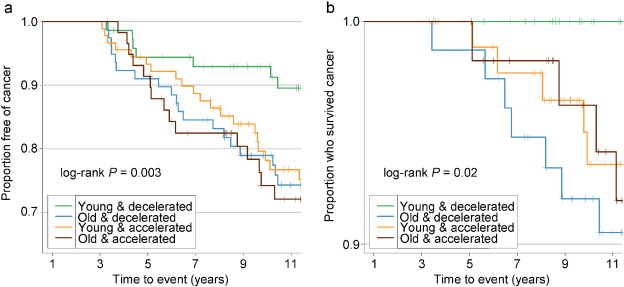
Fig. 3
Kaplan–Meier survival curves comparing risk of cancer grouped by Δage and its rate of change.
A: cancer incidence; B: cancer mortality. Δage slope was calculated for each participant with two or more visits to represent the Δage rate of change. KM survival curves were then plotted among each of the combination of the binary Δage and its rate of change. Young: epigenetically young (Δage ≤ 0); old: epigenetically old (Δage > 0); decelerated: decelerated or stable epigenetic aging over time (Δage slope ≤ 0); accelerated: accelerated epigenetic aging over time (Δage slope > 0).
4. Discussion
Our results indicate a strong, possibly dose-responsive, relationship between Δage and cancer incidence and mortality that is independent of telomere shortening and several epidemiological risk factors for cancer. The relationships between Δage and cancer were strongest in DNA samples collected within three to five years of a cancer event and among participants with accelerated epigenetic aging over time. These findings, coupled with our spline showing a nonlinear, J-shaped relationship between Δage and cancer incidence suggest a dynamic and complex relationship between Δage and cancer over the long term. A diagnostic test to detect an individual's discrepancies between epigenetic and chronological age could therefore prove useful for cancer risk and prognosis assessment.
Epigenetic modifications are potentially central to biological processes of aging (Ben-Avraham et al., 2012), and both global (Ben-Avraham et al., 2012) and gene-specific methylation can be altered substantially by the aging process (Gravina and Vijg, 2010). While the accumulation of epigenetic changes is a hallmark of cancer, few studies have prospectively examined the potential of age-related epigenetic changes to predict cancer. The observed association between Δage and cancer was independent of chronological age, telomere length, and several known lifestyle risk factors for cancer (e.g., body mass index (BMI), and smoking). The method for calculating epigenetic age in our analysis combines age-dependent CpG sites enriched in pathways that are also involved in carcinogenesis (Hannum et al., 2013, Horvath, 2013), and our findings indicate that this may be a viable strategy for developing predictive biomarkers of cancer. In addition, our sensitivity analyses found that the association between Δage and cancer is independent of both telomere length and other comorbidities, suggesting Δage as a specific cancer biomarker as well as the possibility that Δage reflects molecular-level aging or carcinogenic processes that are not captured by telomere measurements.
Our findings are intriguing and biologically plausible. A recent meta-analysis of four large longitudinal cohorts revealed that Δage measured in blood DNA correlates well with all-cause mortality rates and is independent of health, lifestyle, and genetic factors (Marioni et al., 2015). Another recent study of lung cancer found and even stronger relationship between blood epigenetic age and cancer mortality using Horvath's method (Levine et al., 2015). While blood epigenetic age is already being studied in HIV-1 patients with clinical signs of aging (Horvath and Levine, 2015), it may be even more valuable in studies of cancer. WBC play an important role in carcinogenesis via inflammatory response and pro-apoptotic processes (Ichikawa et al., 2011, Schnekenburger et al., 2008), both of which also affect epigenetic age. The discrepancy between epigenetic and chronological age, i.e. Δage, has also been associated with cancer prognosis in tumor tissue data from The Cancer Genome Atlas (Lehne et al., 2015). However, sampling different tissues is not feasible for cancer screening at the population level, both for practical reasons and because different cancers can have tissue-specific, differential, and even opposing effects on epigenetic age (Horvath, 2013, Horvath, 2015), Therefore, our finding of the predictive value of blood-based epigenetic age for cancer risk and mortality is particularly provocative and potentially valuable for use as a screening tool for cancer as it can offer more stable and reliable predictions (Horvath, 2013).
The method used in our study for measuring epigenetic age was based on a model trained on genome-wide DNA methylation extracted from WBC (Hannum et al., 2013). As our measurements were taken from extracted WBC, epigenetic age measured in our study could be a proxy measure for the immune response to cancer initiation and progression, potentially explaining the complex, nonlinear, and time-dependent relationships between Δage, cancer incidence, and cancer mortality we observed. It is well-known that thymic involution with aging can result in diminishing numbers of naïve T cells (Taub and Longo, 2005). Our data also suggested that increasing epigenetic age was associated with loss of naïve CD8+ T cells (r = −0.24, P < 0.001, Supplementary material, Fig. S7). After removing the confounding effects of chronological age and immunosenescence using Δage, our spline plots showed that the risk of developing cancer over time is minimized at Δage = 0, with a more linear risk profile emerging 3–5 years prior to diagnosis (Fig. 2). This may reflect the initial anti-inflammatory and pro-apoptotic response by the body to early carcinogenesis, which triggers increased WBC production that successfully (albeit temporarily) represses cancer. As the cancer eventually adapts to such immune responses they become less effective, resulting in the J-shaped curve evident in our spline plot. Thus, epigenetic age substantially ‘younger’ than chronological age (negative Δage) may be a biomarker of the initial response to a developing cancer before it is detectable through standard diagnostic means. The findings of our Kaplan–Meier curve relating any disruption in the normal epigenetic aging observed in healthy subjects (i.e. either higher Δageor accelerated epigenetic aging over time) to cancer incidence and mortality are also consistent with this hypothesis (Fig. 3, Supplementary material, Fig. S6). As we continue to accrue follow-up data for this cohort, we can assess whether this suggestion is accurate by comparing health outcomes for epigenetically ‘young’ to epigenetically ‘old’ subjects over time. These findings should also be verified in other, larger datasets.
The observed dynamics are also reflected in the straightforward Δage–mortality relationship. Delays between cancer diagnosis and death may allow time for normal thymic involution and other age-related immune deterioration processes to reassert themselves, after the initial response to cancer. This would effectively allow epigenetic age to ‘catch up’ to chronological age and produce the observed linear relationship with mortality. Alternatively, our findings related to cancer mortality could also be reflective of metastasis-induced changes in WBC epigenetic profiles. Longer follow-up in larger groups will be necessary to confirm these hypotheses, but if confirmed, they suggest that Δage would be best utilized as a biomarker of early immune response to carcinogenic processes, with any perturbations of epigenetic age relative to chronological age serving as a potentially useful indicator for use in mass screenings for cancer.
The longitudinal nature of our study enabled us to establish temporal associations between Δage and cancer risk. Our sample size limited our ability to conduct meaningful analyses of cancer subtypes, thus caution should be exercised in interpreting our results. However, while different cancers are biologically distinct from one another blood-based Δage may not be cancer-type-specific. As carcinogenesis usually alters methylation of WBC through inflammation and immune senescence pathways common to most cancer types (Ponnappan and Ponnappan, 2011, Li et al., 2012), epigenetic age acceleration in blood DNA may be a useful biomarker for many (if not all) types of cancer. Therefore, pooling multiple different cancers is biologically plausible for purposes of examining Δage in blood. Additional studies with longer follow-up are needed to confirm this, and to verify the dynamic, nonlinear, and time-dependent relationship between Δage and cancer.
Epigenetic age is comparable to chronological age among cancer-free participants. Those with older epigenetic age relative to their chronological age have an elevated risk of cancer events within three to five years. Our study provides insight into using blood-based epigenetic age as a potential biomarker for cancer early detection. It may also inform future studies of the effects of aging on cancer and potentially other diseases. Expanding this research to additional time points would help determine when in the course of the disease cancer-related changes in epigenetic age occur, and the extent to which Δage is different across individuals and cancers.
Author Contributions
Yinan Zheng, Brian Joyce, and Elena Colicino had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Zheng, Joyce, Hou.
Acquisition, analysis, or interpretation of data: All authors.
Drafting of the manuscript: Zheng, Joyce, Hou.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical analysis: Zheng.
Obtained funding: Hou, Baccarelli, Schwartz.
Administrative, technical, or material support: Jafari.
Study supervision: Hou.
Acknowledgments
The authors thank Alan Aalsburg, Genomics Core Facility, Northwestern University Feinberg School of Medicine, for methylation profiling lab work and raw data generation.
References
- Aryee, M.J., Jaffe, A.E., Corrada-Bravo, H., Ladd-Acosta, C., Feinberg, A.P., Hansen, K.D., and Irizarry, R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014; 30: 1363–1369 DOI: http://dx.doi.org/10.1093/bioinformatics/btu049 (PubMed PMID: 24478339; PubMed Central PMCID: PMC4016708)
- Barfield, R.T., Almli, L.M., Kilaru, V., Smith, A.K., Mercer, K.B., Duncan, R., Klengel, T., Mehta, D., Binder, E.B., Epstein, M.P., Ressler, K.J., and Conneely, K.N. Accounting for population stratification in DNA methylation studies. Genet. Epidemiol. 2014; 38: 231–241 DOI: http://dx.doi.org/10.1002/gepi.2178 (PubMed PMID: 24478250; PubMed Central PMCID: PMC4090102)
- Bell, B., Rose, C.L., and Damon, A. The Veterans Administration longitudinal study of healthy aging. The Gerontologist. 1966; 6: 179–184 (PubMed PMID: 5342911)
- Ben-Avraham, D., Muzumdar, R.H., and Atzmon, G. Epigenetic genome-wide association methylation in aging and longevity. Epigenomics. 2012; 4: 503–509 DOI: http://dx.doi.org/10.2217/epi.12.41 (Epub 2012/11/08, PubMed Central PMCID: PMCPMC4123813)
- Brooks-Wilson, A.R. Genetics of healthy aging and longevity. Hum. Genet. 2013; 132: 1323–1338 (Epub 2013/08/09. doi: 10.1007/s00439-013-1342-z; PubMed Central PMCID: PMCPMC3898394)
- Gravina, S. and Vijg, J. Epigenetic factors in aging and longevity. Pflugers Arch. 2010; 459: 247–258 DOI: http://dx.doi.org/10.1007/s00424-009-0730-7 (Epub 2009/09/22, PubMed PMID: 19768466)
- Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., Klotzle, B., Bibikova, M., Fan, J.B., Gao, Y., Deconde, R., Chen, M., Rajapakse, I., Friend, S., Ideker, T., and Zhang, K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013; 49: 359–367 DOI: http://dx.doi.org/10.1016/j.molcel.2012.10.016 (PubMed PMID: 23177740; PubMed Central PMCID: PMC3780611)
- Hastie, T. and Tibshirani, R. Generalized additive models for medical research. Stat. Methods Med. Res. 1995; 4: 187–196 DOI: http://dx.doi.org/10.1177/096228029500400302 (doi: citeulike-article-id:8292286)
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14: R115 DOI: http://dx.doi.org/10.1186/gb-2013-14-10-r115 (PubMed PMID: 24138928; PubMed Central PMCID: PMC4015143)
- Horvath, S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2015; 16: 96 DOI: http://dx.doi.org/10.1186/s13059-015-0649-6 (PubMed PMID: 25968125; PubMed Central PMCID: PMC4427927)
- Horvath, S. and Levine, A.J. HIV-1 infection accelerates age according to the epigenetic clock. J. Infect. Dis. 2015; DOI: http://dx.doi.org/10.1093/infdis/jiv277 (Epub 2015/05/15)
- Hou, L., Joyce, B.T., Gao, T., Liu, L., Zheng, Y., Penedo, F.J., Liu, S., Zhang, W., Bergan, R., Dai, Q., Vokonas, P., Hoxha, M., Schwartz, J., and Baccarelli, A. Blood telomere length attrition and cancer development in the normative aging study cohort. EBioMedicine. 2015; 2: 591–596 DOI: http://dx.doi.org/10.1016/j.ebiom.2015.04.008
- Houseman, E.A., Accomando, W.P., Koestler, D.C., Christensen, B.C., Marsit, C.J., Nelson, H.H., Wiencke, J.K., and Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 2012; 13: 86 DOI: http://dx.doi.org/10.1186/1471-2105-13-86 (PubMed PMID: 22568884; PubMed Central PMCID: PMC3532182)
- Ichikawa, M., Williams, R., Wang, L., Vogl, T., and Srikrishna, G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol. Cancer Res. 2011; 9: 133–148 DOI: http://dx.doi.org/10.1158/1541-7786.mcr-10-0394 (Epub 2011/01/14, PubMed PMID: 21228116; PubMed Central PMCID: PMCPMC3078037)
- Lehne, B., Drong, A.W., Loh, M., Zhang, W., Scott, W.R., Tan, S.T., Afzal, U., Scott, J., Jarvelin, M.R., Elliott, P., McCarthy, M.I., Kooner, J.S., and Chambers, J.C. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015; 16: 37 DOI: http://dx.doi.org/10.1186/s13059-015-0600-x (PubMed PMID: 25853392; PubMed Central PMCID: PMCPMC4365767)
- Levine, M.E., Hosgood, H.D., Chen, B., Absher, D., Assimes, T., and Horvath, S. DNA methylation age of blood predicts future onset of lung cancer in the women's health initiative. Aging (Albany NY). 2015; 7: 690–700 (PubMed PMID: 26411804; PubMed Central PMCID: PMCPMC4600626)
- Li, L., Choi, J.Y., Lee, K.M., Sung, H., Park, S.K., Oze, I., Pan, K.F., You, W.C., Chen, Y.X., Fang, J.Y., Matsuo, K., Kim, W.H., Yuasa, Y., and Kang, D. DNA methylation in peripheral blood: a potential biomarker for cancer molecular epidemiology. J. Epidemiol. Jpn. Epidemiol. Assoc. 2012; 22: 384–394 (PubMed PMID: 22863985; PubMed Central PMCID: PMC3798632)
- Marioni, R.E., Shah, S., McRae, A.F., Chen, B.H., Colicino, E., Harris, S.E., Gibson, J., Henders, A.K., Redmond, P., Cox, S.R., Pattie, A., Corley, J., Murphy, L., Martin, N.G., Montgomery, G.W., Feinberg, A.P., Fallin, M., Multhaup, M.L., Jaffe, A.E., Joehanes, R., Schwartz, J., Just, A.C., Lunetta, K.L., Murabito, J.M., Starr, J.M., Horvath, S., Baccarelli, A.A., Levy, D., Visscher, P.M., Wray, N.R., and Deary, I.J. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015; 16: 25 DOI: http://dx.doi.org/10.1186/s13059-015-0584-6 (PubMed PMID: 25633388; PubMed Central PMCID: PMC4350614)
- Meira-Machado, L., Cadarso-Suarez, C., Gude, F., and Araujo, A. smoothHR: an R package for pointwise nonparametric estimation of hazard ratio curves of continuous predictors. Comput. Math. Methods Med. 2013; 2013: 745742 DOI: http://dx.doi.org/10.1155/2013/745742 (PubMed PMID: 24454541; PubMed Central PMCID: PMC3876718)
- Plerou, V., Gopikrishnan, P., Rosenow, B., Amaral, L.A., Guhr, T., and Stanley, H.E. Random matrix approach to cross correlations in financial data. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2002; 65: 066126 DOI: http://dx.doi.org/10.1103/PhysRevE.65.066126 (PubMed PMID: 12188802)
- Ponnappan, S. and Ponnappan, U. Aging and immune function: molecular mechanisms to interventions. Antioxid. Redox Signal. 2011; 14: 1551–1585 DOI: http://dx.doi.org/10.1089/ars.2010.3228 (PubMed PMID: 20812785; PubMed Central PMCID: PMC3061194))
- Schnekenburger, J., Schick, V., Kruger, B., Manitz, M.P., Sorg, C., Nacken, W., Kerkhoff, C., Kahlert, A., Mayerle, J., Domschke, W., and Lerch, M.M. The calcium binding protein S100A9 is essential for pancreatic leukocyte infiltration and induces disruption of cell–cell contacts. J. Cell. Physiol. 2008; 216: 558–567 DOI: http://dx.doi.org/10.1002/jcp.21433 (Epub 2008/05/03)
- Taub, D.D. and Longo, D.L. Insights into thymic aging and regeneration. Immunol. Rev. 2005; 205: 72–93 DOI: http://dx.doi.org/10.1111/j.0105-2896.2005.00275.x (PubMed PMID: 15882346)
- Teschendorff, A.E., Zhuang, J., and Widschwendter, M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011; 27: 1496–1505 DOI: http://dx.doi.org/10.1093/bioinformatics/btr171 (PubMed PMID: 21471010)
- Weidner, C.I., Lin, Q., Koch, C.M., Eisele, L., Beier, F., Ziegler, P., Bauerschlag, D.O., Jockel, K.H., Erbel, R., Muhleisen, T.W., Zenke, M., Brummendorf, T.H., and Wagner, W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014; 15: R24 DOI: http://dx.doi.org/10.1186/gb-2014-15-2-r24 (PubMed PMID: 24490752; PubMed Central PMCID: PMC4053864)
- Zhu, Z.Z., Sparrow, D., Hou, L., Tarantini, L., Bollati, V., Litonjua, A.A., Zanobetti, A., Vokonas, P., Wright, R.O., Baccarelli, A., and Schwartz, J. Repetitive element hypomethylation in blood leukocyte DNA and cancer incidence, prevalence, and mortality in elderly individuals: the Normative Aging Study. Cancer Causes Control. 2011; 22: 437–447 DOI: http://dx.doi.org/10.1007/s10552-010-9715-2 (Epub 2010/12/29. PubMed Central PMCID: PMCPMC3752839).